近年來量子化學的發展,已經成功幫助人們解決許多複雜的化學問題。然而,量子化學計算需要大量的運算資源。隨著模擬的系統不斷增大,所需的時間也會大幅提升。因此,改善演算法的運算效率就成為非常重要的研究課題。
對於大部分的量子化學計算來說,結構優化是不可或缺的步驟。常用的二階演算法如擬牛頓法 BFGS可將複雜的目標函數近似為二次函數,在優化的過程中逐漸朝向二次函數的低點移動。然而,當實際目標函數偏離二次函數時,以牛頓法為基礎的二階方法會造成位移過大,甚至會走錯方向。因此,雖然擬牛頓法(如BFGS) 已經被廣泛使用,但它們的收斂速度,可能仍有進步的空間。
為了解決這個問題,我們試著使用強化學習(Reinforcement learning, RL),加快分子結構優化的收斂速度。RL是人工智慧的一個分支,其目標是藉由模型與環境互動,不斷地從錯誤中學習,修正模型所做的決定,以發展出一套最佳的策略完成指定的任務。若要把RL應用在結構優化的問題上,我們可以把優化的過程視為在位能面 (potential energy surface, PES) 上行走,藉著梯度等資訊決定走的方向及步長,試著以最少的步數走到PES的低點。為了達到這個目標,每走一步模型都會得到一次懲罰,因此透過強化學習訓練,模型可以發展出一套策略(π),以最有效率方式走到PES的山谷。如圖一所示,我們成功地結合了RL與擬牛頓法BFGS,開發出高效的分子結構優化演算法 (RL optimizer) ,其收斂速度和傳統的BFGS相比約快了約30% (圖二)。此外,我們也發現RL optimizer的收斂速度非常接近BFGS搭配線搜尋,顯示RL optimizer在位移大小控制上非常出色。然而和BFGS搭配線搜尋相比,RL optimizer不需要額外的梯度運算來達成位移大小的控制,因此可顯著降低理論計算在結構優化上的計算時間。(化工系李奕霈教授提供)
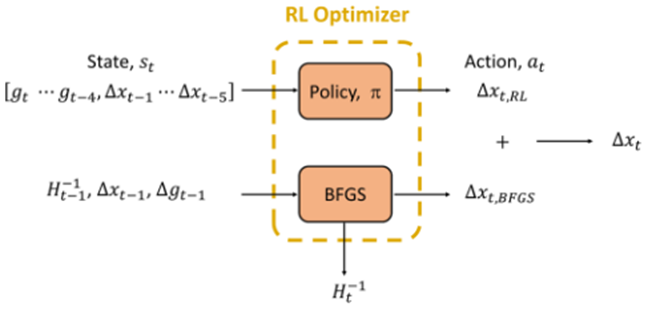
圖一、策略模型(π)和BFGS的關係圖。π的輸出值αt可視為BFGS演算法的修正項。
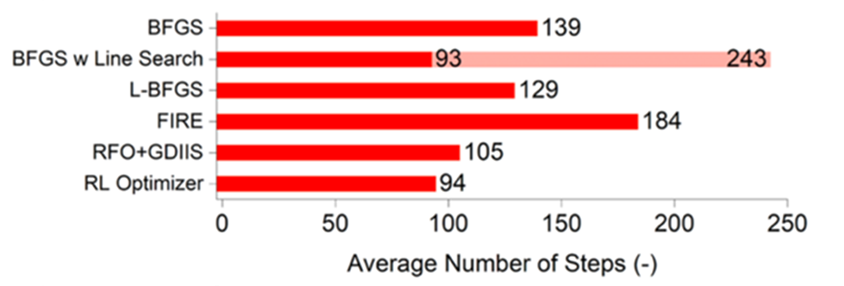
圖二、不同演算法優化分子結構所需的平均步數。BFGS w Line Search顯示的陰影條,代表線搜索所產生的額外梯度計算。