奈米孔洞材料例如金屬有機骨架(metal-organic frameworks,MOFs)近年來引起了非常大的關注,它們具有作為氣體分離技術中關鍵材料的許多理想性質例如高吸附選擇性和大表面積。這類材料的另一個關鍵特徵為它們的結構可調性,例如金屬有機骨架通過不同金屬與有機配位基的組合,迄今為止已經有超過10,000個結構被成功合成,並還存在數個量級多的可能結構,雖然如此多樣的材料結構為不同應用提供了無限的可能,卻也讓最佳材料的開發與尋找極具挑戰性。
雖然使用大規模分子模擬來協助尋找最佳材料已經取得非常大的進展,直接採用分子模擬篩選材料仍然需要耗費大量電腦資源。因此,近幾年來機器學習(machine learning, ML)技術被視為一個非常有潛力的方法,藉由建立ML模型來迅速且準確地預測材料之性質。傳統的方式為使用一些結構特徵(features)來描述結構並基於這些特徵來訓練一個機器學習模型於性質的預測,這些特徵可能包含結構之最大孔洞直徑、孔隙率等等。然而,此類方式所建立的模型之準確率往往直接取決於所選用的"特徵”。 我們的研究提出不同的做法,利用新穎的convolutional neural network(CNN)模型來訓練機器直接“看”結構,並從而減少人為偏差。如圖一所示,基於三維孔洞結構內每個原子的位子,使用每個原子的化學性質包含其Lennard-Jones參數(ϵ與σ)和電荷q作為該位子的特徵值,然後最佳化convolutional layer並利用fully connected neural networks以進行性質預測。我們的結果也展現此方法具有非常大的潛力,如圖二所示,利用這方法所建立的3維CNN模型可以預測甲烷與帶有quadrupole moment的二氧化碳在MOFs裡的吸附性質,特別是ㄧ般ML模型非常困難描述的無限稀釋條件下的亨利吸附常數。(化工系林立強教授提供)
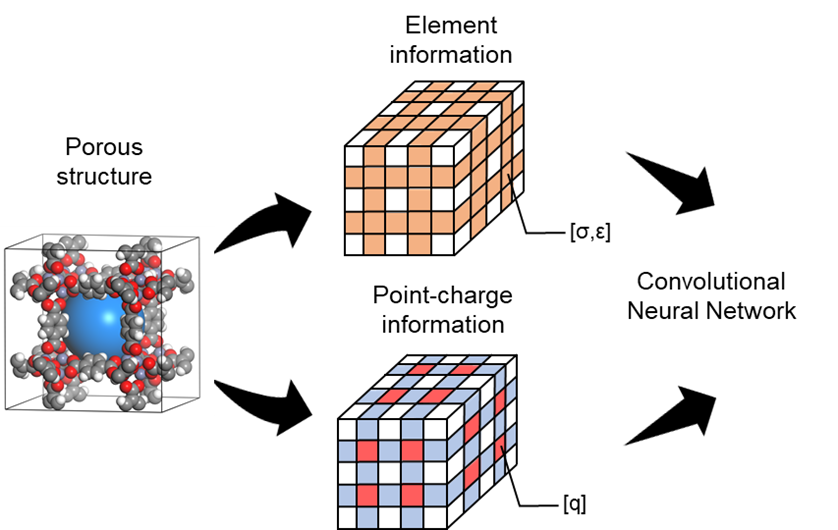
圖一: 使用結構內每個原子的化學性值包含其Lennard-Jones參數(ϵ與σ)和電荷q作為特徵值來建立convolutional neural network(CNN)模型。此圖節錄於Hung et al., J. Phys. Chem. C 2022, 126, 5, 2813–2822.
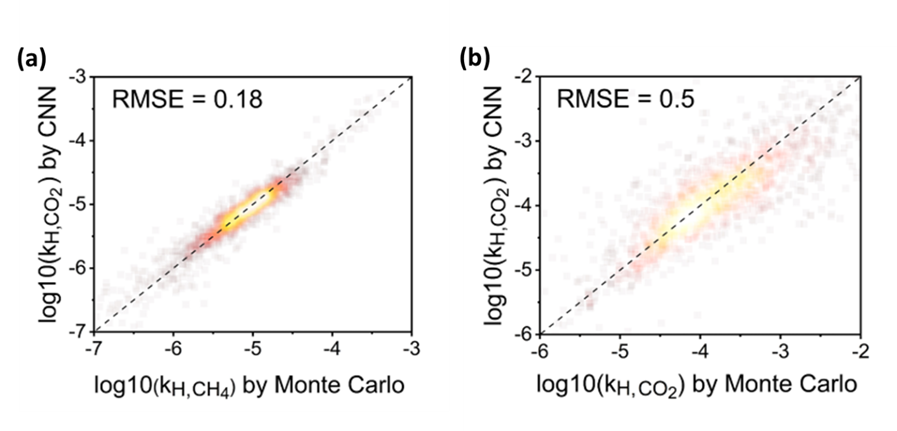
圖二: CNN模型所預測之(a)甲烷和(b)二氧化碳於MOFs內無限稀釋條件下的亨利吸附常數與蒙地卡羅(Monte Carlo) 所計算的參考數值之比較。此圖節錄於Hung et al., J. Phys. Chem. C 2022, 126, 5, 2813–2822.